Packaging and storage—
Preserve in well-closed containers. Store between 15
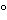
and 30
, or under refrigeration.
Chromatographic purity—
Diluting solution—
Prepare a solution of butylated hydroxytoluene in acetonitrile containing 0.5 mg per mL.
Standard solutions—
Dissolve an accurately weighed quantity of
USP Simvastatin RS in
Diluting solution to obtain
Standard solution A having a known concentration of about 0.2 mg per mL. Transfer 4.0, 2.0, and 1.0 mL of
Standard solution A to separate 10-mL volumetric flasks, and dilute with
Diluting solution to volume to obtain
Standard solutions B, C, and
D, respectively.
Test solution—
Dissolve an accurately weighed quantity of simvastatin in Diluting solution to obtain a solution having a concentration of about 20 mg per mL.
Procedure—
Separately apply 4-µL portions of each of the
Standard solutions and the
Test solution to a thin-layer chromatographic plate (see
Chromatography  621
621
) coated with a 0.25-mm layer of silica gel mixture, previously washed with methanol and air-dried. Dry the spots with the aid of a stream of nitrogen. Position the plate in a chromatographic chamber previously equilibrated with a solvent system consisting of a mixture of cyclohexane, chloroform, and isopropyl alcohol (5:2:1) containing 0.5 mg of butylated hydroxytoluene per mL, and develop the chromatogram until the solvent front has moved about three-quarters of the length of the plate. Remove the plate from the developing chamber, mark the solvent front, and allow the solvent to evaporate under a stream of nitrogen. Spray the plate with a mixture of methanol and sulfuric acid (8:2), heat at 110
for 30 minutes, and immediately examine the plate: no secondary spot in the chromatogram of the
Test solution is greater in size or intensity than the principal spot from
Standard solution B (0.4%), and the sum of all such secondary spots obtained from the
Test solution is not greater than 1.0%.
Limit of lovastatin—
From the chromatograms of the
Assay preparation and the
Standard preparation, obtained as directed in the
Assay, calculate the percentage of lovastatin in the portion of Simvastatin taken by the formula:
10,000(C / W)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Lovastatin RS in the
Standard preparation; W is the weight, in mg, of Simvastatin taken for the
Assay preparation; and
rU and
rS are the lovastatin peak responses obtained from the
Assay preparation and the
Standard preparation, respectively: not more than 1% is found.
Assay—
Mobile phase—
Prepare a filtered and degassed mixture of acetonitrile and dilute phosphoric acid (1 in 1000) (50:50). Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Diluting solution—
Prepare a mixture of acetonitrile and 0.01 M monobasic potassium phosphate (60:40), filter, and adjust with phosphoric acid to a pH of 4.0.
Assay preparation—
Transfer about 30 mg of Simvastatin, accurately weighed, to a 100-mL volumetric flask, and dissolve in and dilute with Diluting solution to volume.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 238-nm detector and a 4.6- × 33-mm column that contains packing L1. The flow rate is about 3.0 mL per minute. Chromatograph the
Standard preparation, and record the peak responses as directed for
Procedure: the relative retention times are about 0.65 for lovastatin and 1.0 for simvastatin; the resolution,
R, between simvastatin and lovastatin is not less than 3.0; the column efficiency is not less than 2000 theoretical plates; the tailing factor is not more than 2.0; and the relative standard deviation for replicate injections is not more than 1.0%.
Procedure—
Separately inject equal volumes (about 5 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the areas of the responses for the major peaks. Calculate the quantity, in mg, of C
25H
38O
5 in the portion of Simvastatin taken by the formula:
100C(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Simvastatin RS in the
Standard preparation; and
rU and
rS are the peak responses obtained from the
Assay preparation and the
Standard preparation, respectively.
;)